
STANDING UP TO SICKLE CELL ANEMIA
Introduction
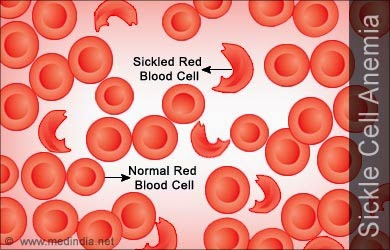
Sickle cell disease (also called sickle cell anemia) is an inherited blood disorder that affects red blood cells. The sickle cell gene causes the body to produce abnormal hemoglobin. In sickle cell disease, the hemoglobin clumps together, causing red blood cells to become stiff and develop a C-shaped (“sickle”) form. These sickled red blood cells can block blood vessels, reducing blood flow in many parts of the body. This process results in tissue and organ damage.
Hemoglobin and Iron
Each red blood cell contains about 280 million hemoglobin molecules. Hemoglobin is the most important component of red blood cells. It is composed of protein (globulin) and a molecule (heme), which binds to iron.
In the lungs, the heme component takes up oxygen and releases carbon dioxide. The red blood cells carry the oxygen to the body’s tissues, where the hemoglobin releases the oxygen in exchange for carbon dioxide, and the cycle repeats. The oxygen is essential for all cells in the body to function.
(adsbygoogle = window.adsbygoogle || []).push({});
Sickle cell disease reduces or denies adequate oxygen to many parts of the body. This contributes to the severe pain experienced as a sickle cell crisis and both short- and long-term organ damage.
Causes
Sickle Cell Disease and Hemoglobin
Sickle cell disease occurs from genetic changes that cause abnormalities in hemoglobin molecules:
Hemoglobin A (HbA):HbA is the hemoglobin molecule found in normal red blood cells during childhood and adulthood. People who do not have sickle cell disease generally have this type of hemoglobin in their blood cells.
Hemoglobin S (HbS):HbS (S is for sickle) is the abnormal variant of hemoglobin A, which occurs in sickle-red blood cells and is the primary problem in the disease. The difference between hemoglobin A (HbA) and hemoglobin S (HbS) is only one protein out of about 300 that are common to both. This protein lies along an amino-acid chain called beta-globin, where even a tiny abnormality has serious consequences.
Hemoglobin is the most important component of red blood cells. It is composed of a protein called heme, which binds oxygen. In the lungs, oxygen is exchanged for carbon dioxide. Abnormalities of an individual’s hemoglobin value can indicate defects in red blood cell balance. Both low and high values can indicate disease states.
The Sickle Cell Disease Process
(adsbygoogle = window.adsbygoogle || []).push({});
The symptoms and problems of sickle cell disease are a result of the hemoglobin S (HbS) molecule:
When the sickle hemoglobin molecule loses its oxygen, it forms rigid rods called polymers that change the red blood cells into a sickle or crescent shape. These sickle-shaped cells stick to the walls and cannot squeeze through the capillaries. Blood flow through tiny blood vessels becomes slowed or stopped in many parts of the body. This deprives tissues and organs of oxygen. When this blood flow slows or stops suddenly in a certain part of the body, the decrease in oxygen (hypoxia) can cause severe pain (the sickle cell crisis). Over time, it leads to gradual destruction in organs and tissues throughout the body.
Diet Significance:
There is no cure for sickle cell disease; however, a balanced diet consisting of healthy food options with adequate nutrient content may prevent painful complications such as a sickle cell crisis, according to FamilyDoctor.org. A sickle cell crisis is defined as a sharp pain throughout the body that may last several hours to several days due to formation of blood clots. Individuals diagnosed with sickle cell anemia can continue to live productive lives if they manage their pain by adopting a healthy lifestyle.
Sickle Cell Disease: Nutritional Considerations

Patients with sickle cell disease have increased needs for calories and micronutrients (e.g., vitamins and minerals). A diet emphasizing fruits, vegetables, whole grains, and legumes will provide a greater proportion of essential nutrients than a typical Western diet, and appropriate supplementation (one to three times the recommended intakes for most essential nutrients) can prevent deficiency.
A high–calorie, Nutrient–dense Diet:
The average caloric intake of sickle cell patients is typically low, especially during flare–ups of the disease. As a result, children with sickle cell disease have impaired growth and significantly lower weight compared with unaffected individuals. A careful nutritional assessment and the addition of energy supplements are needed.
Adequate fluid consumption to maintain hydration:
Sickling of red blood cells increases when patients become dehydrated. Thus, it is important to maintain hydration by consuming adequate fluids. In some cases, hospitalization to receive intravenous fluids may become necessary.
Vitamin and mineral supplementation: Blood levels of several vitamins and minerals are often low in individuals with sickle cell disease, including vitamin A and carotenoids, vitamin B6, vitamin C, vitamin E, magnesium, and zinc. This can result in a significant deficiency of antioxidants, which may increase the risk of disease flare–ups. Studies indicate that vitamin–mineral supplements of certain nutrients (vitamins C and E, zinc, and magnesium) or treatment with a combination of high–dose antioxidants can reduce the percentage of sickled red blood cells.
Omega–3 Fatty Acid Supplements:
Supplementation with omega–3 fatty acids can improve the membranes of red blood cells and may decrease flare–ups of the disease. A small preliminary study indicated that omega–3 fatty acid supplementation with fish oil reduced the number of painful episodes requiring hospitalization. Some studies claim that omega-three fatty acids, found in fish and soybean oil as well as dietary supplements, might make red blood cell membranes less fragile and possibly less likely to sickle, although no studies have proven this definitively.
Folic acid:
The requirement for folic acid in sickle cell disease is higher than in people without the disease. This increased requirement is caused by the increased rate of production of red blood cells that occurs in sickle cell disease. Some clinics recommend use of folic acid, 5mg once a day, but a normal balanced diet usually contains sufficient folic acid and a daily supplement is generally not required but if it is taken it does no harm.
Take Steps To Prevent and Control Complications

Along with adopting healthy lifestyle habits, steps should be taken to prevent and control painful sickle cell crises. Many factors can cause sickle cell crises. Knowing how to avoid or control these factors can help manage pain.
*Avoid decongestants, such as pseudoephedrine:
These medicines can tighten blood vessels, making it harder for red blood cells to move smoothly through the vessels.
*Abstain from Too hot or too cold Temperatures:
Extremes of temperature and a sudden change from a warm to a cold environment can trigger a sickle cell crisis. When a person is cold in order to keep the blood warm and flowing the walls of the blood vessels thickens making the space in the middle of the blood vessel smaller. This is not a problem if the red blood cells are a normal shape and the blood is not too thick however if the cells are sickle shaped, hard and rigid and the blood is thick the cells are more likely to cause an obstruction and sickling crisis.
Conversely if a person is too hot they will sweat more and lose more water from the body, when this happens the blood will become thicker. In order to prevent the body from becoming dehydrated the kidneys will reduce the amount of urine it is producing and will make more concentrated urine. The kidney of people with sickle cell disease is not able to concentrate urine efficiently therefore even when they are too hot the kidney continues to produce large amounts of diluted urine and this leads to dehydration and cause increasing sickling which can lead to a sickle cell crisis. Therefore it is important to avoid extremes of temperatures, wrapping up and wearing sufficiently warm clothing when it is cold and avoiding chilling is essential. During hot whether it is necessary to keep the body cool and well hydrated.
*Excessive physical stress:

During physical exercise the body needs extra oxygen in order to provide for active muscles. People with sickle cell disease have a chronic anaemia which reduces their ability to carry oxygen very well therefore during physical exercise they become breathless easily and get tired quickly.
Although it is important to maintain physical activity this should be done in moderation. Every person is different whilst some people with sickle cell disease can withstand a moderate amount of physical exertion others cannot. Each individual will need to identify their own physical tolerance level and maintain their own physical boundary.
Physical stress includes those things that place a greater physical burden on the body for example pregnancy, it is not uncommon for women to experience more frequent sickling crisis during pregnancy and in the early weeks after delivery of the baby.
Definition and Causes of Sickle Cell AnemiaFactors a Sickler Must ConsiderHome Remedies for Sickle Cell Anaemia




{kind=link}